Cluster analysis of CC DD overlap DE genes FC#
CC/C DD/D overlap differential expression overlap gene foldchange cluster in CC/C and DD/D
[1]:
library(UpSetR)
library(readxl)
library(ggplot2)
library(clusterProfiler)
library(ClusterGVis)
library(Mfuzz)
library(AnnotationHub)
library(biomaRt)
library(ComplexHeatmap)
library(dplyr)
library(stringr)
clusterProfiler v3.16.1 For help: https://guangchuangyu.github.io/software/clusterProfiler
If you use clusterProfiler in published research, please cite:
Guangchuang Yu, Li-Gen Wang, Yanyan Han, Qing-Yu He. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS: A Journal of Integrative Biology. 2012, 16(5):284-287.
Attaching package: ‘clusterProfiler’
The following object is masked from ‘package:stats’:
filter
Loading required package: Biobase
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: ‘BiocGenerics’
The following objects are masked from ‘package:parallel’:
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from ‘package:stats’:
IQR, mad, sd, var, xtabs
The following objects are masked from ‘package:base’:
anyDuplicated, append, as.data.frame, basename, cbind, colnames,
dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget,
order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply,
union, unique, unsplit, which, which.max, which.min
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: e1071
Warning message in fun(libname, pkgname):
“no display name and no $DISPLAY environment variable”
Attaching package: ‘DynDoc’
The following object is masked from ‘package:BiocGenerics’:
path
Attaching package: ‘Mfuzz’
The following object is masked from ‘package:ClusterGVis’:
filter.std
Loading required package: BiocFileCache
Loading required package: dbplyr
Attaching package: ‘AnnotationHub’
The following object is masked from ‘package:Biobase’:
cache
Loading required package: grid
========================================
ComplexHeatmap version 2.15.1
Bioconductor page: http://bioconductor.org/packages/ComplexHeatmap/
Github page: https://github.com/jokergoo/ComplexHeatmap
Documentation: http://jokergoo.github.io/ComplexHeatmap-reference
If you use it in published research, please cite either one:
- Gu, Z. Complex Heatmap Visualization. iMeta 2022.
- Gu, Z. Complex heatmaps reveal patterns and correlations in multidimensional
genomic data. Bioinformatics 2016.
The new InteractiveComplexHeatmap package can directly export static
complex heatmaps into an interactive Shiny app with zero effort. Have a try!
This message can be suppressed by:
suppressPackageStartupMessages(library(ComplexHeatmap))
========================================
Attaching package: ‘dplyr’
The following object is masked from ‘package:biomaRt’:
select
The following objects are masked from ‘package:dbplyr’:
ident, sql
The following object is masked from ‘package:widgetTools’:
funs
The following object is masked from ‘package:Biobase’:
combine
The following objects are masked from ‘package:BiocGenerics’:
combine, intersect, setdiff, union
The following objects are masked from ‘package:stats’:
filter, lag
The following objects are masked from ‘package:base’:
intersect, setdiff, setequal, union
[220]:
[2]:
hub <- AnnotationHub() #建立AnnotationHub对象(视人品,网不行加载不了)
# unique(hub$species) #查看AnonotationHub里面物种
hub$species[which(hub$species=="Solanum")] #看AnonotationHub里是否包含想要的物种
# Solanum是番茄的拉丁名
query(hub, "Solanum") #查看该物种信息
hub[hub$species=="Solanum" & hub$rdataclass == "OrgDb"] #OrgDb属于rdataclass中,因此查看下该物种有没有OrgDb
Solanum.OrgDb <- hub[["AH80808"]]#AH59087是番茄对应的编号
using temporary cache /var/folders/s6/8xqxsbh11nl91f72l5vsz7fh0000gn/T//RtmpciKBs5/BiocFileCache
snapshotDate(): 2020-04-27
AnnotationHub with 8 records
# snapshotDate(): 2020-04-27
# $dataprovider: ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/, Inparanoid8
# $species: Solanum tuberosum, Solanum lycopersicum, Solanum pennellii, Sola...
# $rdataclass: OrgDb, Inparanoid8Db
# additional mcols(): taxonomyid, genome, description,
# coordinate_1_based, maintainer, rdatadateadded, preparerclass, tags,
# rdatapath, sourceurl, sourcetype
# retrieve records with, e.g., 'object[["AH10593"]]'
title
AH10593 | hom.Solanum_lycopersicum.inp8.sqlite
AH10606 | hom.Solanum_tuberosum.inp8.sqlite
AH80691 | org.Solanum_pennelli.eg.sqlite
AH80692 | org.Solanum_pennellii.eg.sqlite
AH80747 | org.Solanum_tuberosum.eg.sqlite
AH80807 | org.Solanum_esculentum.eg.sqlite
AH80808 | org.Solanum_lycopersicum.eg.sqlite
AH80809 | org.Solanum_lycopersicum_var._humboldtii.eg.sqlite
AnnotationHub with 0 records
# snapshotDate(): 2020-04-27
downloading 1 resources
retrieving 1 resource
loading from cache
Loading required package: AnnotationDbi
Loading required package: stats4
Loading required package: IRanges
Loading required package: S4Vectors
Attaching package: ‘S4Vectors’
The following object is masked from ‘package:clusterProfiler’:
rename
The following object is masked from ‘package:base’:
expand.grid
Attaching package: ‘IRanges’
The following object is masked from ‘package:clusterProfiler’:
slice
Attaching package: ‘AnnotationDbi’
The following object is masked from ‘package:clusterProfiler’:
select
[6]:
CC_FC <- read.table("CC_overlap_log2fc.csv", header=TRUE, row.names=1, sep="\t")
colnames(CC_FC) <- paste0("HAG", gsub("^([[:alpha:]]*).[[:alpha:]].", '', colnames(CC_FC)))
DD_FC <- read.table("DD_overlap_log2fc.csv", header=TRUE, row.names=1, sep="\t")
colnames(DD_FC) <- paste0("HAG", gsub("^([[:alpha:]]*).[[:alpha:]].", '', colnames(DD_FC)))
[13]:
geneIDTransForm <- read.table("~/Documents/phd/tomato_metabolic/geneIDTransForm/mart_export.txt", sep="\t", header=TRUE)
[67]:
id2entrezID <- function(ids){
#df <- geneIDTransForm[geneIDTransForm$NCBI.gene..formerly.Entrezgene..ID,]
Entreids <- unique(geneIDTransForm[geneIDTransForm$Gene.stable.ID %in% ids,]$NCBI.gene..formerly.Entrezgene..ID)
return(Entreids)
}
cluster_gene_go <- function(entirz, clusteri){
cluster_erich.go = enrichGO(gene = entirz,
OrgDb = Solanum.OrgDb,
keyType = "ENTREZID",
ont = "BP",
pvalueCutoff = 0.5,
qvalueCutoff = 0.5)
df <- cluster_erich.go@result
df$Group <- clusteri
df <- subset(df, select = c("Group", "Description", "pvalue", "GeneRatio"))
colnames(df) <- c("group", "Description", "pvalue", "ratio")
return (df)
}
cm2go <- function(cm_n){
clusters <- unique(cm_n$wide.res$cluster)
all_cluster_go <- data.frame()
for (cl in clusters){
sub_cm_genes <- subset(cm_n$wide.res, cluster==cl, select=c("gene"))
sub_cm_entriz <- id2entrezID(sub_cm_genes$gene)
cluster_go <- cluster_gene_go(sub_cm_entriz, paste0("C", cl))
if (dim(all_cluster_go)[1]==0){
all_cluster_go <- cluster_go
}else{
all_cluster_go = rbind(all_cluster_go, cluster_go)
}
}
return (all_cluster_go)
}
[ ]:
CC#
[283]:
mfuzz_cluster_num = 18
CC_FC_cm <- clusterData(exp = CC_FC,
cluster.method = "mfuzz",
cluster.num = mfuzz_cluster_num)
CC_FC_cluster_go <- cm2go(CC_FC_cm)
saveRDS(CC_FC_cm, paste0("mfuzz_data/CC_FC_cm_cluster",mfuzz_cluster_num,".rds"))
saveRDS(CC_FC_cluster_go, paste0("mfuzz_data/CC_FC_cluster_go_cluster",mfuzz_cluster_num,".rds"))
0 genes excluded.
[2]:
mfuzz_cluster_num = 18
CC_FC_cm <- readRDS(paste0("mfuzz_data/CC_FC_cm_cluster",mfuzz_cluster_num,".rds"))
CC_FC_cluster_go <- readRDS(paste0("mfuzz_data/CC_FC_cluster_go_cluster",mfuzz_cluster_num,".rds"))
CC_topGO <- CC_FC_cluster_go %>% group_by(group) %>% slice_head(n = 6)
CC_topGO <- data.frame(CC_topGO)
[4]:
#write.table(CC_FC_cluster_go, "CC_FC_cluster_go.csv", sep=",", row.names=TRUE)
[ ]:
[2]:
#write.table(CC_FC_cm$wide.res, "CC_FC_cm_mfuzz.csv", sep=",", row.names=FALSE)
[ ]:
[276]:
#subset(CC_FC_cluster_go, Description=="regulation of macromolecule biosynthetic process")
[12]:
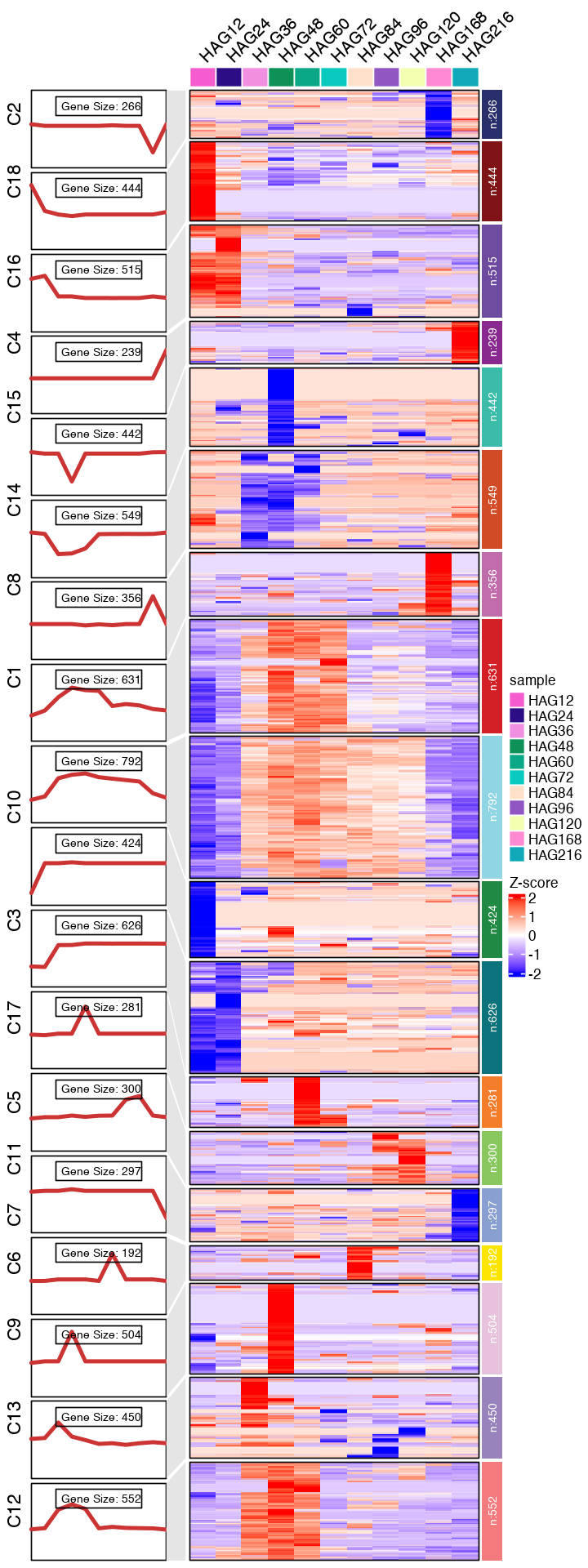
options(repr.plot.width=6, repr.plot.height=16)
#pdf(paste0("mfuzz_cluster_fig/CC_mfuzz_cluster_", mfuzz_cluster_num,".pdf"), width = 12, height = 20, onefile = T)
CC_topGO <- CC_FC_cluster_go %>% group_by(group) %>% slice_head(n = 6)
CC_topGO <- data.frame(CC_topGO)
visCluster(object = CC_FC_cm,
plot.type = "both",
column_names_rot = 45,
show_row_dend = F,
#markGenes = markGenes,
markGenes.side = "left",
genes.gp = c('italic',fontsize = 12,col = "black"),
#annoTerm.data = subset(CC_topGO, select=c("group", "Description", "pvalue")),
line.side = "left",
#go.col = rep(ggsci::pal_d3()(mfuzz_cluster_num),each = 7),
#go.col = rep(ggsci::pal_d3("category20")(mfuzz_cluster_num),each = 6),
#go.size = "pval",
#add.bar = T,
#subgroup.anno = c("C4","C6","C8")
)
#dev.off()
[1] "This palatte have 20 colors!"
[8]:
options(repr.plot.width=12, repr.plot.height=20)
#pdf(paste0("mfuzz_cluster_fig/CC_mfuzz_cluster_", mfuzz_cluster_num,".pdf"), width = 12, height = 20, onefile = T)
CC_topGO <- CC_FC_cluster_go %>% group_by(group) %>% slice_head(n = 6)
CC_topGO <- data.frame(CC_topGO)
visCluster(object = CC_FC_cm,
plot.type = "both",
column_names_rot = 45,
show_row_dend = F,
#markGenes = markGenes,
markGenes.side = "left",
genes.gp = c('italic',fontsize = 12,col = "black"),
annoTerm.data = subset(CC_topGO, select=c("group", "Description", "pvalue")),
line.side = "left",
#go.col = rep(ggsci::pal_d3()(mfuzz_cluster_num),each = 7),
go.col = rep(ggsci::pal_d3("category20")(mfuzz_cluster_num),each = 6),
#go.size = "pval",
add.bar = T,
#subgroup.anno = c("C4","C6","C8")
)
#dev.off()
[1] "This palatte have 20 colors!"
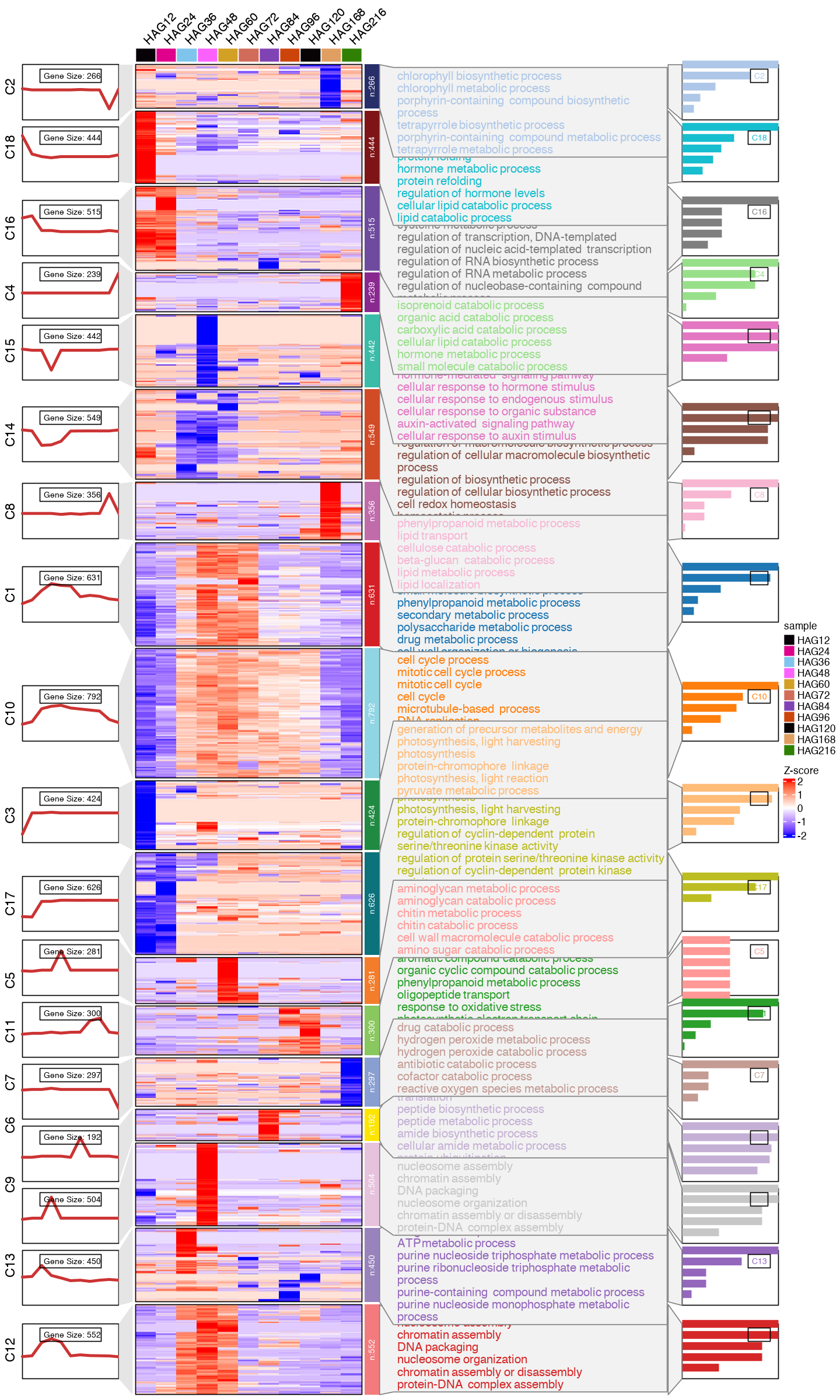
[23]:
#c2show <- c("C12", "C1", "C10", "C3", "C17", "C14", "C15", "C2", "C7")
#c2showi <- c(12, 1, 10, 3, 17, 14, 15, 2, 7)
c2show <- c("C18", "C16", "C13", "C9", "C5", "C6", "C11", "C8", "C4")
c2showi <- c(18, 16, 13, 9, 5, 6, 11, 8, 4)
CC_FC_cm2show <- CC_FC_cm
CC_FC_cm2show$wide.res <- subset(CC_FC_cm2show$wide.res, cluster %in% c2showi)
CC_FC_cm2show$wide.res$cluster <- factor(CC_FC_cm2show$wide.res$cluster, levels=c2showi)
CC_FC_cm2show$long.res <- subset(CC_FC_cm2show$long.res, cluster %in% c2showi)
CC_FC_cm2show$long.res$cluster <- factor(CC_FC_cm2show$long.res$cluster, levels=c2showi)
CC_topGO2show <- subset(CC_topGO, group%in% c2show)
CC_topGO2show$group <- factor(CC_topGO2show$group, levels=c2show)
cl.info <- data.frame(table(CC_FC_cm2show$wide.res$cluster)) %>%
dplyr::arrange(Var1)
cluster.num <- nrow(cl.info)
subgroup <- lapply(1:nrow(cl.info),function(x){
nm <- rep(as.character(cl.info$Var1[x]),cl.info$Freq[x])
paste("C",nm,sep = '')
}) %>% unlist()
align_to = split(1:nrow(CC_FC_cm2show$wide.res), subgroup)
align_to = align_to[paste0("C", cl.info$Var1)]
[ ]:
mat <- CC_FC_cm2show$wide.res
#mat <- mat[order(mat$cluster, mat$cluster),]
mat <- mat[order(mat$cluster),]
mat <- mat %>%
#dplyr::arrange(cluster) %>%
dplyr::select(-gene,-cluster,-membership)
ComplexHeatmap::Heatmap(mat,
name = "Z-score",
cluster_rows = FALSE,
cluster_columns = FALSE,
show_row_names = FALSE,
#column_names_side = "top",
row_split = factor(subgroup, levels=names(align_to))
)
[322]:
align_to = split(1:nrow(CC_FC_cm2show$wide.res), subgroup)
[24]:
options(repr.plot.width=12, repr.plot.height=10)
#pdf(paste0("mfuzz_cluster_fig/CC_mfuzz_cluster_", mfuzz_cluster_num,"_mainFig.pdf"), width = 12, height = 10, onefile = T)
visCluster2(object = CC_FC_cm2show,
plot.type = "both",
column_names_rot = 45,
show_row_dend = F,
border=TRUE,
#markGenes = markGenes,
markGenes.side = "left",
genes.gp = c('italic',fontsize = 12,col = "black"),
annoTerm.data = subset(CC_topGO2show, select=c("group", "Description", "pvalue")),
line.side = "left",
#go.col = rep(ggsci::pal_d3()(mfuzz_cluster_num),each = 7),
go.col = rep(ggsci::pal_d3("category20")(length(c2show)),each = 6),
#go.size = "pval",
add.bar = T,
textbar.pos = c(0.8,0.2),
#subgroup.anno = c2show,
#subgroup.anno = c("C4","C6","C8")
)
#dev.off()
[1] "C18" "C16" "C13" "C9" "C5" "C6" "C11" "C8" "C4"
[1] 18 16 13 9 5 6 11 8 4
Levels: 18 16 13 9 5 6 11 8 4
[1] "This palatte have 20 colors!"
[1] "C18" "C16" "C13" "C9" "C5" "C6" "C11" "C8" "C4"
[1] 90909090
[1] 8
[1] 12121212121
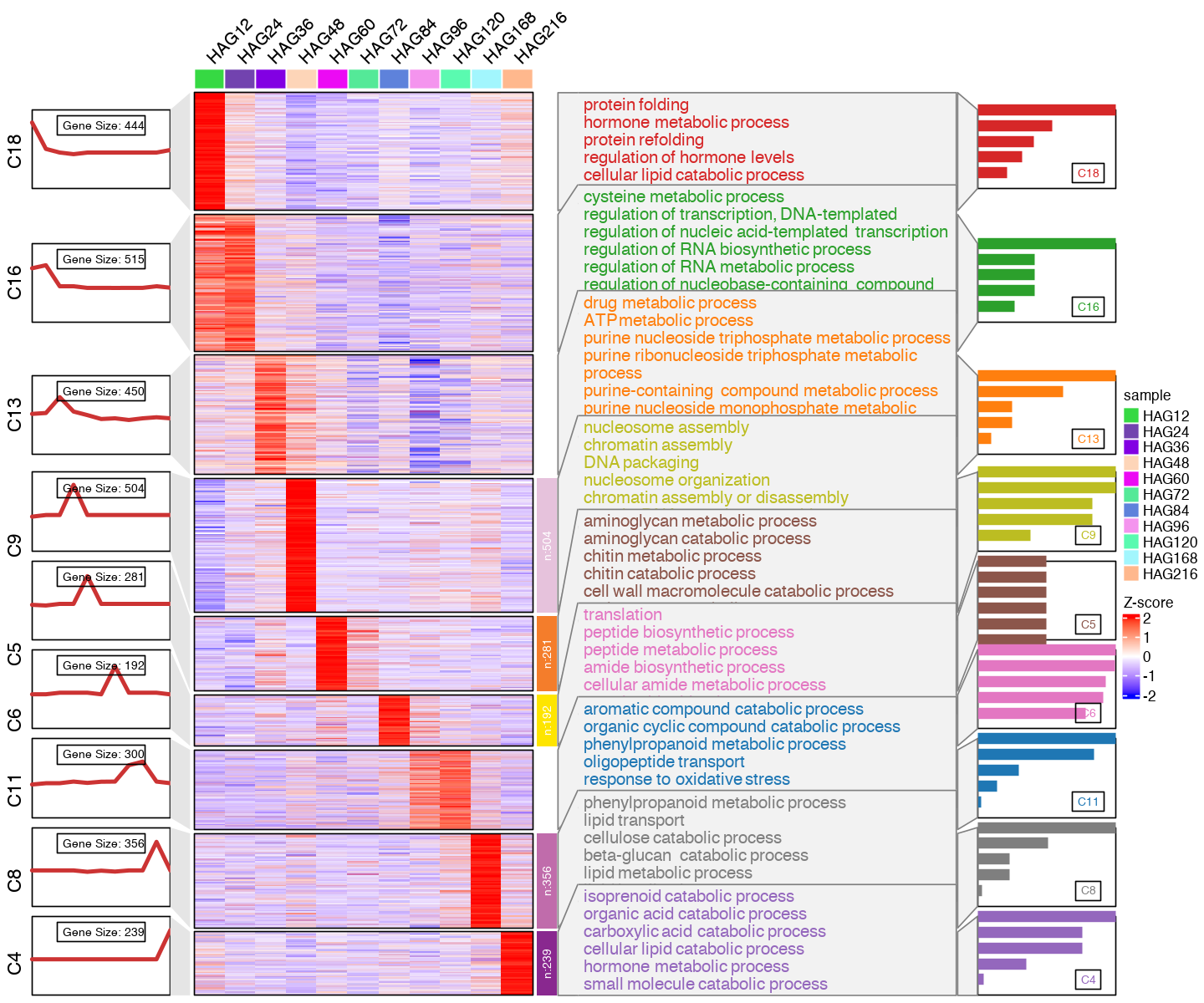
DD for paper#
[ ]:
[10]:
#c2show <- c("C5", "C18", "C6", "C15", "C11", "C9", "C12", "C10")
#c2showi <- c(5, 18, 6, 15, 11, 9, 12, 10)
c2show <- c("C3", "C13", "C16", "C8", "C14", "C2", "C1", "C4", "C17", "C7")
c2showi <- c(3, 13, 16, 8, 14, 2, 1, 4, 17, 7)
CC_FC_cm2show <- DD_FC_cm
CC_FC_cm2show$wide.res <- subset(CC_FC_cm2show$wide.res, cluster %in% c2showi)
CC_FC_cm2show$wide.res$cluster <- factor(CC_FC_cm2show$wide.res$cluster, levels=c2showi)
CC_FC_cm2show$long.res <- subset(CC_FC_cm2show$long.res, cluster %in% c2showi)
CC_FC_cm2show$long.res$cluster <- factor(CC_FC_cm2show$long.res$cluster, levels=c2showi)
CC_topGO2show <- subset(DD_topGO, group%in% c2show)
CC_topGO2show$group <- factor(CC_topGO2show$group, levels=c2show)
options(repr.plot.width=12, repr.plot.height=10)
pdf(paste0("mfuzz_cluster_fig/DD_mfuzz_cluster_", mfuzz_cluster_num,"_suppFig.pdf"), width = 12, height = 10, onefile = T)
visCluster2(object = CC_FC_cm2show,
plot.type = "both",
column_names_rot = 45,
show_row_dend = F,
border=TRUE,
#markGenes = markGenes,
markGenes.side = "left",
genes.gp = c('italic',fontsize = 12,col = "black"),
annoTerm.data = subset(CC_topGO2show, select=c("group", "Description", "pvalue")),
line.side = "left",
#go.col = rep(ggsci::pal_d3()(mfuzz_cluster_num),each = 7),
go.col = rep(ggsci::pal_d3("category20")(length(c2show)),each = 6),
#go.size = "pval",
add.bar = T,
textbar.pos = c(0.8,0.2),
#subgroup.anno = c2show,
#subgroup.anno = c("C4","C6","C8")
)
dev.off()
Error in eval(expr, envir, enclos): 找不到对象'DD_FC_cm'
Traceback:
[ ]:
[366]:
CC_FC_cm2show$type
'mfuzz'
[13]:
globalVariables(c('cell_type', 'cluster.num', 'gene',"ratio","bary",
'membership', 'norm_value','id', 'log10P', 'pval',
'Var1'))
visCluster2 <- function(object = NULL,
# plot.data = NULL,
ht.col = c("blue", "white", "red"),
border = TRUE,
plot.type = c("line","heatmap","both"),
ms.col = c('#0099CC','grey90','#CC3333'),
line.size = 0.1,
line.col = "grey90",
add.mline = TRUE,
mline.size = 2,
mline.col = "#CC3333",
ncol = 4,
ctAnno.col = NULL,
set.md = "median",
textbox.pos = c(0.5,0.8),
textbox.size = 8,
# panel size,gap,width,fill,col
panel.arg = c(2,0.25,4,"grey90",NA),
annoTerm.data = NULL,
annoTerm.mside = "right",
# textbox fill and col
termAnno.arg = c("grey95","grey50"),
add.box = FALSE,
boxcol = NULL,
# box with and border color
box.arg = c(0.1,"grey50"),
add.point = FALSE,
# shape,fill,color,size
point.arg = c(19,"orange","orange",1),
add.line = TRUE,
line.side = "right",
markGenes = NULL,
markGenes.side = "right",
genes.gp = c('italic',10,NA),
go.col = NULL,
go.size = NULL,
term.text.limit = c(10,18),
mulGroup = NULL,
lgd.label = NULL,
show_row_names = FALSE,
subgroup.anno = NULL,
add.bar = FALSE,
bar.width = 8,
textbar.pos = c(0.8,0.8),
annnoblock.text = TRUE,
annnoblock.gp = c("white",8),
add.sampleanno = TRUE,
sample.group = NULL,
sample.col = NULL,
sample.order = NULL,
HeatmapAnnotation = NULL,
column.split = NULL,
...){
ComplexHeatmap::ht_opt(message = FALSE)
col_fun = circlize::colorRamp2(c(-2, 0, 2), ht.col)
plot.type <- match.arg(plot.type)
# choose plot type
if(plot.type == "both"){
# ==========================================================================
# process data
# if(is.null(plot.data)){
# data <- data.frame(object$wide.res)
# }else{
# data <- plot.data
# }
data <- data.frame(object$wide.res)
# prepare matrix
if(object$type == "mfuzz"){
#mat <- data %>%
# dplyr::arrange(cluster) %>%
# dplyr::select(-gene,-cluster,-membership)
#mat <- data
mat <- data[order(object$wide.res$cluster),]
mat <- mat %>%
#dplyr::arrange(cluster) %>%
dplyr::select(-gene,-cluster,-membership)
rownames(mat) <- data$gene
# sample orders
if(!is.null(sample.order)){
mat <- mat[,sample.order]
}
# split info
cl.info <- data.frame(table(data$cluster)) %>%
dplyr::arrange(Var1)
cluster.num <- nrow(cl.info)
subgroup <- lapply(1:nrow(cl.info),function(x){
nm <- rep(as.character(cl.info$Var1[x]),cl.info$Freq[x])
paste("C",nm,sep = '')
}) %>% unlist()
print(unique(subgroup))
print(cl.info$Var1)
subgroup <- factor(subgroup, levels=paste0("C",cl.info$Var1))
# plot
# =================== bar annotation for samples
# sample group info
if(is.null(sample.group)){
sample.info = colnames(mat)
# split columns
if(is.null(HeatmapAnnotation)){
column_split = NULL
}else{
column_split = column.split
}
}
# order
sample.info <- factor(sample.info,levels = unique(sample.info))
# sample colors
if(is.null(sample.col)){
# scol <- ggsci::pal_npg()(length(sample.info))
scol <- circlize::rand_color(n = length(sample.info))
names(scol) <- sample.info
}else{
scol <- sample.col
names(scol) <- sample.info
}
# top anno
if(add.sampleanno == TRUE){
if(is.null(HeatmapAnnotation)){
topanno = ComplexHeatmap::HeatmapAnnotation(sample = sample.info,
col = list(sample = scol),
gp = grid::gpar(col = "white"),
show_annotation_name = FALSE)
}else{
topanno = HeatmapAnnotation
}
}else{
topanno = NULL
}
# =================== bar annotation for clusters
if(is.null(ctAnno.col)){
colanno <- jjAnno::useMyCol("stallion",n = cluster.num)
}else{
colanno <- ctAnno.col
}
names(colanno) <- 1:cluster.num
# anno.block <- ComplexHeatmap::anno_block(gp = grid::gpar(fill = colanno,col = NA),
# which = "row")
align_to = split(1:nrow(mat), subgroup)
#print(444444)
#print(align_to)
align_to = align_to[paste0("C", cl.info$Var1)]
print(names(align_to))
#print(55555)
#print(align_to)
#print(str(align_to))
anno.block <- ComplexHeatmap::anno_block(align_to = align_to,
panel_fun = function(index, nm) {
npos = as.numeric(unlist(strsplit(nm,split = "C"))[2])
# rect
grid::grid.rect(gp = grid::gpar(fill = colanno[npos],col = NA))
# text
if(annnoblock.text == TRUE){
grid::grid.text(label = paste("n:",length(index),sep = ''),
rot = 90,
gp = grid::gpar(col = annnoblock.gp[1],
fontsize = as.numeric(annnoblock.gp[2])))
}
},
which = "row")
# =================== gene annotation for heatmap
# whether mark your genes on plot
if(!is.null(markGenes)){
# all genes
rowGene <- rownames(mat)
# tartget gene
annoGene <- markGenes
# add color for gene
gene.col <- data %>%
dplyr::select(gene,cluster) %>%
dplyr::filter(gene %in% annoGene)
purrr::map_df(1:cluster.num,function(x){
tmp <- gene.col %>%
dplyr::filter(cluster == x) %>%
dplyr::mutate(col = colanno[x])
}) -> gene.col
gene.col <- gene.col[match(annoGene,gene.col$gene),]
if(is.na(genes.gp[3])){
gcol = gene.col$col
}else{
gcol = genes.gp[3]
}
# get target gene index
index <- match(annoGene,rowGene)
# some genes annotation
geneMark = gene = ComplexHeatmap::anno_mark(at = index,
labels = annoGene,
which = "row",
side = markGenes.side,
labels_gp = grid::gpar(fontface = genes.gp[1],
fontsize = as.numeric(genes.gp[2]),
col = gcol))
}else{
geneMark = NULL
}
# final annotation for heatmap
right_annotation = ComplexHeatmap::rowAnnotation(gene = geneMark,cluster = anno.block)
# =======================================================
# return plot according to plot type
if(plot.type == "both"){
#====================== heatmap + line
rg = range(mat)
# # panel_fun for line plot
# panel_fun = function(index, nm) {
# grid::pushViewport(grid::viewport(xscale = c(1,ncol(mat)), yscale = rg))
# grid::grid.rect()
#
# # grid.xaxis(gp = gpar(fontsize = 8))
# # grid.annotation_axis(side = 'right',gp = gpar(fontsize = 8))
#
# # choose method
# if(set.md == "mean"){
# mdia <- colMeans(mat[index, ])
# }else if(set.md == "median"){
# mdia <- apply(mat[index, ], 2, stats::median)
# }else{
# print("supply mean/median !")
# }
#
# # get gene numbers
# text <- paste("Gene Size:",nrow(mat[index, ]),sep = ' ')
# ComplexHeatmap::grid.textbox(text,x = textbox.pos[1],y = textbox.pos[2],
# gp = grid::gpar(fontsize = textbox.size,fontface = "italic"))
#
# # grid.points(x = 1:ncol(m),y = mdia,
# # pch = 19,
# # gp = gpar(col = 'orange'))
#
# grid::grid.lines(x = scales::rescale(1:ncol(mat),to = c(0,1)),
# y = scales::rescale(mdia,to = c(0,1),from = rg),
# gp = grid::gpar(lwd = 3,col = mline.col))
#
# grid::popViewport()
# }
# ====================================================================
# panel_fun for line plot
panel_fun = function(index, nm) {
# whether add boxplot
if(add.box == TRUE & add.line != TRUE){
xscale = c(-0.1,1.1)
}else{
xscale = c(0,1)
}
grid::pushViewport(grid::viewport(xscale = xscale, yscale = c(0,1)))
grid::grid.rect()
# grid.xaxis(gp = gpar(fontsize = 8))
# grid.annotation_axis(side = 'right',gp = gpar(fontsize = 8))
# # choose method
# if(set.md == "mean"){
# mdia <- colMeans(mat[index, ])
# }else if(set.md == "median"){
# mdia <- apply(mat[index, ], 2, stats::median)
# }else{
# print("supply mean/median !")
# }
#
# # boxplot xpos
# pos = scales::rescale(1:ncol(mat),to = c(0,1))
#
# # boxcol
# if(is.null(boxcol)){
# boxcol <- rep("grey90",ncol(mat))
# }else{
# boxcol <- boxcol
# }
#
# # boxplot grobs
# if(add.box == TRUE){
# lapply(1:ncol(mat), function(x){
# ComplexHeatmap::grid.boxplot(scales::rescale(mat[index, ][,x],
# to = c(0,1),
# from = c(rg[1] - 0.5,rg[2] + 0.5)),
# pos = pos[x],
# direction = "vertical",
# box_width = as.numeric(box.arg[1]),
# outline = FALSE,
# gp = grid::gpar(col = box.arg[2],fill = boxcol[x]))
# })
# }
#
# # points grobs
# if(add.point == TRUE){
# grid::grid.points(x = scales::rescale(1:ncol(mat),to = c(0,1)),
# y = scales::rescale(mdia,to = c(0,1),from = c(rg[1] - 0.5,rg[2] + 0.5)),
# pch = as.numeric(point.arg[1]),
# gp = grid::gpar(fill = point.arg[2],col = point.arg[3]),
# size = grid::unit(as.numeric(point.arg[4]), "char"))
# }
#
# # lines grobs
# if(add.line == TRUE){
# grid::grid.lines(x = scales::rescale(1:ncol(mat),to = c(0,1)),
# y = scales::rescale(mdia,to = c(0,1),from = c(rg[1] - 0.5,rg[2] + 0.5)),
# gp = grid::gpar(lwd = 3,col = mline.col))
# }
# whether given multiple groups
if(is.null(mulGroup)){
mulGroup <- ncol(mat)
# ================ calculate group columns index
seqn <- data.frame(st = 1,
sp = ncol(mat))
}else{
mulGroup <- mulGroup
grid::grid.lines(x = c(0,1),y = rep(0.5,2),
gp = grid::gpar(col = "black",lty = "dashed"))
# ================ calculate group columns index
cu <- cumsum(mulGroup)
seqn <- data.frame(st = c(1,cu[1:(length(cu) - 1)] + 1),
sp = c(cu[1],cu[2:length(cu)]))
}
# loop for multiple groups to create grobs
lapply(1:nrow(seqn), function(x){
tmp <- seqn[x,]
tmpmat <- mat[index, c(tmp$st:tmp$sp)]
# choose method
if(set.md == "mean"){
mdia <- colMeans(tmpmat)
}else if(set.md == "median"){
mdia <- apply(tmpmat, 2, stats::median)
}else{
print("supply mean/median !")
}
# boxplot xpos
pos = scales::rescale(1:ncol(tmpmat),to = c(0,1))
# boxcol
if(is.null(boxcol)){
boxcol <- rep("grey90",ncol(tmpmat))
}else{
boxcol <- boxcol
}
# boxplot grobs
if(add.box == TRUE){
lapply(1:ncol(tmpmat), function(x){
ComplexHeatmap::grid.boxplot(scales::rescale(tmpmat[,x],
to = c(0,1),
from = c(rg[1] - 0.5,rg[2] + 0.5)),
pos = pos[x],
direction = "vertical",
box_width = as.numeric(box.arg[1]),
outline = FALSE,
gp = grid::gpar(col = box.arg[2],fill = boxcol[x]))
})
}
# points grobs
if(add.point == TRUE){
grid::grid.points(x = scales::rescale(1:ncol(tmpmat),to = c(0,1)),
y = scales::rescale(mdia,to = c(0,1),from = c(rg[1] - 0.5,rg[2] + 0.5)),
pch = as.numeric(point.arg[1]),
gp = grid::gpar(fill = point.arg[2],col = point.arg[3]),
size = grid::unit(as.numeric(point.arg[4]), "char"))
}
# lines grobs
if(add.line == TRUE){
grid::grid.lines(x = scales::rescale(1:ncol(tmpmat),to = c(0,1)),
y = scales::rescale(mdia,to = c(0,1),from = c(rg[1] - 0.5,rg[2] + 0.5)),
gp = grid::gpar(lwd = 3,col = mline.col[x]))
}
})
# get gene numbers
grid.textbox <- utils::getFromNamespace("grid.textbox", "ComplexHeatmap")
text <- paste("Gene Size:",nrow(mat[index, ]),sep = ' ')
grid.textbox(text,x = textbox.pos[1],y = textbox.pos[2],
gp = grid::gpar(fontsize = textbox.size,
fontface = "italic",
...))
grid::popViewport()
}
# whether annotate subgroups
if(!is.null(subgroup.anno)){
align_to = split(1:nrow(mat), subgroup)
align_to = align_to[paste0("C", cl.info$Var1)]
align_to = align_to[subgroup.anno]
#subgroup <- factor(subgroup, levels=names(align_to))
}else{
##### align_to = subgroup
align_to = split(1:nrow(mat), subgroup)
align_to = align_to[paste0("C", cl.info$Var1)]
#subgroup <- factor(subgroup, levels=names(align_to))
}
# anno link annotation
anno = ComplexHeatmap::anno_link(align_to = align_to,
which = "row",
panel_fun = panel_fun,
size = grid::unit(as.numeric(panel.arg[1]), "cm"),
gap = grid::unit(as.numeric(panel.arg[2]), "cm"),
width = grid::unit(as.numeric(panel.arg[3]), "cm"),
side = line.side,
link_gp = grid::gpar(fill = panel.arg[4],col = panel.arg[5]))
# =====================================
# whether add go term annotations
if(!is.null(annoTerm.data)){
# load term info
termanno <- annoTerm.data
if(ncol(termanno) == 2){
colnames(termanno) <- c("id","term")
}else if(ncol(termanno) == 3){
colnames(termanno) <- c("id","term","pval")
}else if(ncol(termanno) == 4){
colnames(termanno) <- c("id","term","pval","ratio")
}else{
print("No more than 4 columns!")
}
# term colors
if(is.null(go.col)){
gocol <- circlize::rand_color(n = nrow(termanno))
}else{
gocol <- go.col
}
# term text size
if(is.null(go.size)){
gosize <- rep(12,nrow(termanno))
}else{
if(go.size == "pval"){
# loop for re-scaling pvalue
purrr::map_df(unique(termanno$id),function(x){
tmp <- termanno %>%
dplyr::filter(id == x) %>%
dplyr::mutate(size = scales::rescale(-log10(pval),to = term.text.limit))
}) -> termanno.tmp
gosize <- termanno.tmp$size
}else{
gosize <- go.size
}
}
# add to termanno
termanno <- termanno %>%
dplyr::mutate(col = gocol,fontsize = gosize)
# to list
lapply(1:length(unique(termanno$id)), function(x){
tmp = termanno[which(termanno$id == unique(termanno$id)[x]),]
df <- data.frame(text = tmp$term,
col = tmp$col,
fontsize = tmp$fontsize)
return(df)
}) -> term.list
# add names
names(term.list) <- unique(termanno$id)
# whether annotate subgroups
if(!is.null(subgroup.anno)){
align_to2 = split(seq_along(subgroup), subgroup)
align_to = align_to[paste0("C", cl.info$Var1)]
align_to2 = align_to2[subgroup.anno]
#subgroup <- factor(subgroup, levels=names(align_to))
term.list = term.list[subgroup.anno]
}else{
align_to2 = subgroup
#align_to2 = align_to2[paste0("C", cl.info$Var1)]
term.list = term.list
}
# textbox annotations
# if(add.bar == TRUE){
# box.side = "left"
# }else{
# box.side = "right"
# }
textbox = ComplexHeatmap::anno_textbox(align_to2, term.list,
word_wrap = TRUE,
add_new_line = TRUE,
side = annoTerm.mside,
background_gp = grid::gpar(fill = termAnno.arg[1],
col = termAnno.arg[2]))
# final row annotation
# if(line.side == "right"){
# right_annotation2 = ComplexHeatmap::rowAnnotation(cluster = anno.block,
# line = anno,
# textbox = textbox)
# left_annotation = NULL
# }else{
# right_annotation2 = ComplexHeatmap::rowAnnotation(cluster = anno.block,
# textbox = textbox)
# left_annotation = ComplexHeatmap::rowAnnotation(line = anno)
# }
# GO bar anno function
if(ncol(termanno) - 2 > 2){
anno_gobar <- function(data = NULL,
bar.width = 0.1,
# col = NA,
align_to = NULL,
panel.arg = panel.arg,
...){
# process data
if(ncol(data) - 2 == 3){
data <- data %>%
dplyr::mutate(bary = -log10(pval))
}else{
data <- data %>%
dplyr::mutate(bary = ratio)
}
ComplexHeatmap::anno_zoom(align_to = align_to,
which = "row",
# =====================
panel_fun = function(index,nm){
grid::pushViewport(grid::viewport(xscale = c(0,1),yscale = c(0,1)))
grid::grid.rect()
# sub data
tmp <- data %>%
dplyr::filter(id == nm) %>%
dplyr::arrange(bary)
# bar grobs
# grid::grid.rect(x = rep(0,nrow(tmp)),
# y = scales::rescale(1:nrow(tmp),to = c(0,1)),
# width = scales::rescale(tmp$log10P,to = c(0,1)),
# height = bar.width,
# gp = grid::gpar(fill = tmp$col,col = col))
grid::grid.segments(x0 = rep(0,nrow(tmp)),
x1 = scales::rescale(tmp$bary,to = c(0,1)),
y0 = scales::rescale(1:nrow(tmp),to = c(0,1)),
y1 = scales::rescale(1:nrow(tmp),to = c(0,1)),
gp = grid::gpar(lwd = bar.width,
col = tmp$col,
lineend = "butt"))
# add cluster name
grid.textbox <- utils::getFromNamespace("grid.textbox", "ComplexHeatmap")
text <- nm
grid.textbox(text,
x = textbar.pos[1],y = textbar.pos[2],
gp = grid::gpar(fontsize = textbox.size,
fontface = "italic",
col = unique(tmp$col),
...))
grid::popViewport()
},
# =======================
size = grid::unit(as.numeric(panel.arg[1]), "cm"),
gap = grid::unit(as.numeric(panel.arg[2]), "cm"),
width = grid::unit(as.numeric(panel.arg[3]), "cm"),
side = "right",
link_gp = grid::gpar(fill = termAnno.arg[1],col = termAnno.arg[2]),
...)
}
# ================================
# bar anno
#print(align_to2)
print(90909090)
print(bar.width)
baranno = anno_gobar(data = termanno,
align_to = align_to2,
panel.arg = panel.arg,
bar.width = bar.width)
}
# whether add bar annotation
if(add.bar == TRUE){
baranno
}else{
baranno = NULL
}
}else{
# ======================================================
# no GO annotation
# if(line.side == "right"){
# right_annotation2 = ComplexHeatmap::rowAnnotation(cluster = anno.block,line = anno)
# left_annotation = NULL
# }else{
# right_annotation2 = ComplexHeatmap::rowAnnotation(cluster = anno.block)
# left_annotation = ComplexHeatmap::rowAnnotation(line = anno)
# }
textbox = NULL
baranno = NULL
}
# ====================================================
# final row annotations
if(line.side == "right"){
if(markGenes.side == "right"){
right_annotation2 = ComplexHeatmap::rowAnnotation(gene = geneMark,
cluster = anno.block,
line = anno,
textbox = textbox,
bar = baranno)
left_annotation = NULL
}else{
right_annotation2 = ComplexHeatmap::rowAnnotation(cluster = anno.block,
line = anno,
textbox = textbox,
bar = baranno)
left_annotation = ComplexHeatmap::rowAnnotation(gene = geneMark)
}
}else{
if(markGenes.side == "right"){
right_annotation2 = ComplexHeatmap::rowAnnotation(gene = geneMark,
cluster = anno.block,
textbox = textbox,
bar = baranno)
left_annotation = ComplexHeatmap::rowAnnotation(line = anno)
}else{
right_annotation2 = ComplexHeatmap::rowAnnotation(cluster = anno.block,
textbox = textbox,
bar = baranno)
left_annotation = ComplexHeatmap::rowAnnotation(line = anno,
gene = geneMark)
}
}
# save
# pdf('test.pdf',height = 10,width = 10)
#str(subgroup)
#print(tail(mat))
print(12121212121)
htf <- ComplexHeatmap::Heatmap(as.matrix(mat),
name = "Z-score",
cluster_columns = FALSE,
cluster_rows = FALSE,
show_row_names = show_row_names,
border = border,
column_split = column_split,
top_annotation = topanno,
right_annotation = right_annotation2,
left_annotation = left_annotation,
column_names_side = "top",
row_split = subgroup,
col = col_fun,
...)
# draw lines legend
if(is.null(mulGroup)){
ComplexHeatmap::draw(htf,merge_legend = TRUE)
}
# dev.off()
}
}
}
}
- 'cell_type'
- 'cluster.num'
- 'gene'
- 'ratio'
- 'bary'
- 'membership'
- 'norm_value'
- 'id'
- 'log10P'
- 'pval'
- 'Var1'
[ ]:
[ ]:
DD#
[475]:
mfuzz_cluster_num = 18
DD_FC_cm <- clusterData(exp = DD_FC,
cluster.method = "mfuzz",
cluster.num = mfuzz_cluster_num)
DD_FC_cluster_go <- cm2go(DD_FC_cm)
saveRDS(DD_FC_cm, paste0("mfuzz_data/DD_FC_cm_cluster",mfuzz_cluster_num,".rds"))
saveRDS(DD_FC_cluster_go, paste0("mfuzz_data/DD_FC_cluster_go_cluster",mfuzz_cluster_num,".rds"))
DD_topGO <- DD_FC_cluster_go %>% group_by(group) %>% slice_head(n = 6)
DD_topGO <- data.frame(DD_topGO)
0 genes excluded.
[5]:
mfuzz_cluster_num = 18
DD_FC_cm <- readRDS(paste0("mfuzz_data/DD_FC_cm_cluster",mfuzz_cluster_num,".rds"))
DD_FC_cluster_go <- readRDS(paste0("mfuzz_data/DD_FC_cluster_go_cluster",mfuzz_cluster_num,".rds"))
DD_topGO <- DD_FC_cluster_go %>% group_by(group) %>% slice_head(n = 6)
DD_topGO <- data.frame(DD_topGO)
Error in DD_FC_cluster_go %>% group_by(group) %>% slice_head(n = 6): 没有"%>%"这个函数
Traceback:
[6]:
#write.table(DD_FC_cm$wide.res, "DD_FC_cm_mfuzz.csv", sep=",", row.names=FALSE)
[8]:
#write.table(DD_FC_cluster_go, "DD_FC_cluster_go.csv", sep=",", row.names=TRUE)
[ ]:
[4]:
options(repr.plot.width=14, repr.plot.height=20)
#pdf(paste0("mfuzz_cluster_fig/DD_mfuzz_cluster_", mfuzz_cluster_num,".pdf"), width = 12, height = 20, onefile = T)
visCluster(object = DD_FC_cm,
plot.type = "both",
column_names_rot = 45,
show_row_dend = F,
#markGenes = markGenes,
markGenes.side = "left",
genes.gp = c('italic',fontsize = 12,col = "black"),
annoTerm.data = subset(DD_topGO, select=c("group", "Description", "pvalue")),
line.side = "left",
#go.col = rep(ggsci::pal_d3()(mfuzz_cluster_num),each = 7),
go.col = rep(ggsci::pal_d3("category20")(mfuzz_cluster_num),each = 6),
go.size = "pval",
add.bar = T,
#subgroup.anno = c("C4","C6","C8")
)
#dev.off()
[1] "This palatte have 20 colors!"

[ ]:
[ ]:
[ ]:
[ ]:
[ ]: