Do heatmap1-2#
[2]:
library(UpSetR)
library(readxl)
library(ggplot2)
library(clusterProfiler)
library(ClusterGVis)
library(Mfuzz)
library(AnnotationHub)
library(biomaRt)
library(ComplexHeatmap)
library(dplyr)
library(stringr)
library(circlize)
library(pheatmap)
clusterProfiler v3.16.1 For help: https://guangchuangyu.github.io/software/clusterProfiler
If you use clusterProfiler in published research, please cite:
Guangchuang Yu, Li-Gen Wang, Yanyan Han, Qing-Yu He. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS: A Journal of Integrative Biology. 2012, 16(5):284-287.
Attaching package: ‘clusterProfiler’
The following object is masked from ‘package:stats’:
filter
Loading required package: Biobase
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: ‘BiocGenerics’
The following objects are masked from ‘package:parallel’:
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from ‘package:stats’:
IQR, mad, sd, var, xtabs
The following objects are masked from ‘package:base’:
anyDuplicated, append, as.data.frame, basename, cbind, colnames,
dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget,
order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply,
union, unique, unsplit, which, which.max, which.min
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: e1071
Warning message in fun(libname, pkgname):
“no display name and no $DISPLAY environment variable”
Attaching package: ‘DynDoc’
The following object is masked from ‘package:BiocGenerics’:
path
Attaching package: ‘Mfuzz’
The following object is masked from ‘package:ClusterGVis’:
filter.std
Loading required package: BiocFileCache
Loading required package: dbplyr
Attaching package: ‘AnnotationHub’
The following object is masked from ‘package:Biobase’:
cache
Loading required package: grid
========================================
ComplexHeatmap version 2.15.1
Bioconductor page: http://bioconductor.org/packages/ComplexHeatmap/
Github page: https://github.com/jokergoo/ComplexHeatmap
Documentation: http://jokergoo.github.io/ComplexHeatmap-reference
If you use it in published research, please cite either one:
- Gu, Z. Complex Heatmap Visualization. iMeta 2022.
- Gu, Z. Complex heatmaps reveal patterns and correlations in multidimensional
genomic data. Bioinformatics 2016.
The new InteractiveComplexHeatmap package can directly export static
complex heatmaps into an interactive Shiny app with zero effort. Have a try!
This message can be suppressed by:
suppressPackageStartupMessages(library(ComplexHeatmap))
========================================
Attaching package: ‘dplyr’
The following object is masked from ‘package:biomaRt’:
select
The following objects are masked from ‘package:dbplyr’:
ident, sql
The following object is masked from ‘package:widgetTools’:
funs
The following object is masked from ‘package:Biobase’:
combine
The following objects are masked from ‘package:BiocGenerics’:
combine, intersect, setdiff, union
The following objects are masked from ‘package:stats’:
filter, lag
The following objects are masked from ‘package:base’:
intersect, setdiff, setequal, union
========================================
circlize version 0.4.15
CRAN page: https://cran.r-project.org/package=circlize
Github page: https://github.com/jokergoo/circlize
Documentation: https://jokergoo.github.io/circlize_book/book/
If you use it in published research, please cite:
Gu, Z. circlize implements and enhances circular visualization
in R. Bioinformatics 2014.
This message can be suppressed by:
suppressPackageStartupMessages(library(circlize))
========================================
Attaching package: ‘pheatmap’
The following object is masked from ‘package:ComplexHeatmap’:
pheatmap
plot heatmap1#
high correlation with sugar genes in grafting stages, heatmap view these genes expression in HAG samples
[6]:
df <- read.table("heatmap1.csv", sep="\t", header=TRUE, row.names=1)
df_meta <- read.table("heatmap1_meta.csv", sep="\t", header=TRUE, row.names=2)
df_meta <- df_meta[,c('Condition', 'HAG')]
df_meta$Condition <- factor(df_meta$Condition, levels=c('C', 'D', 'CC', 'DD', 'CD'))
[1]:
options(repr.plot.width=6, repr.plot.height=10)
#pdf('heatmap1.pdf', width = 12, height=10)
ann_colors = list(
HAG = c("white", "firebrick"),
Condition = c(C = "#E0CCD2", D = "#07CCD2", CC = "#E0CC71", DD = "#8AA6CE", CD = "#8AA69C")
)
pheatmap(log2(df+0.01), annotation_col = df_meta, annotation_colors=ann_colors, border_color=NA,
cluster_rows=TRUE, cluster_cols=FALSE,
show_rownames=TRUE, show_colnames=FALSE,
#color = colorRampPalette(c("navy","white","firebrick3"))(256),
color = colorRampPalette(c("blue", "white", "yellow"))(256),
scale = "row",
width=10, height=12,) #filename="heatmap1.pdf")
#dev.off()
Error in pheatmap(log2(df + 0.01), annotation_col = df_meta, annotation_colors = ann_colors, : 没有"pheatmap"这个函数
Traceback:
plot heatmap2#
high correlation with sugar TFs in grafting stages, heatmap view these TFs expression in HAG samples
[9]:
df2 <- read.table("heatmap2.csv", sep="\t", header=TRUE, row.names=1)
df_meta2 <- read.table("heatmap1_meta.csv", sep="\t", header=TRUE, row.names=2)
df_meta2 <- df_meta2[,c('Condition', 'HAG')]
df_meta2$Condition <- factor(df_meta2$Condition, levels=c('C', 'D', 'CC', 'DD', 'CD'))
options(repr.plot.width=6, repr.plot.height=2)
#pdf('heatmap1.pdf', width = 12, height=10)
ann_colors = list(
HAG = c("white", "firebrick"),
Condition = c(C = "#E0CCD2", D = "#07CCD2", CC = "#E0CC71", DD = "#8AA6CE", CD = "#8AA69C")
)
pheatmap(log2(df2+0.01), annotation_col = df_meta2, annotation_colors=ann_colors, border_color=NA,
cluster_rows=TRUE, cluster_cols=FALSE,
show_rownames=TRUE, show_colnames=FALSE,
color = colorRampPalette(c("navy","white","firebrick3"))(256),
scale = "row", fontsize=8,
width=6, height=2,)
#filename="heatmap2.pdf")
#dev.off()
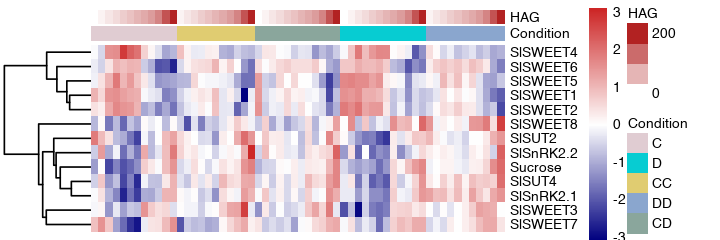
another view type#
[47]:
ha1 = HeatmapAnnotation(df = df_meta,
col = list(Condition = c("C" = "#E0CCD2", "D" = "#07CCD2", "CC" = "#E0CC71", "DD" = "#8AA6CE", "CD" = "#8AA69C"),
HAG = colorRamp2(c(0, 100), c("white", "red")))
)
Heatmap(log2(df+0.1), top_annotation = ha1,
cluster_columns=FALSE, cluster_rows=FALSE,
show_row_names=FALSE, show_column_names=FALSE)
Warning message:
“The input is a data frame-like object, convert it to a matrix.”

[ ]: